Medical device labeling requirements: FDA, MDR & UDI guide
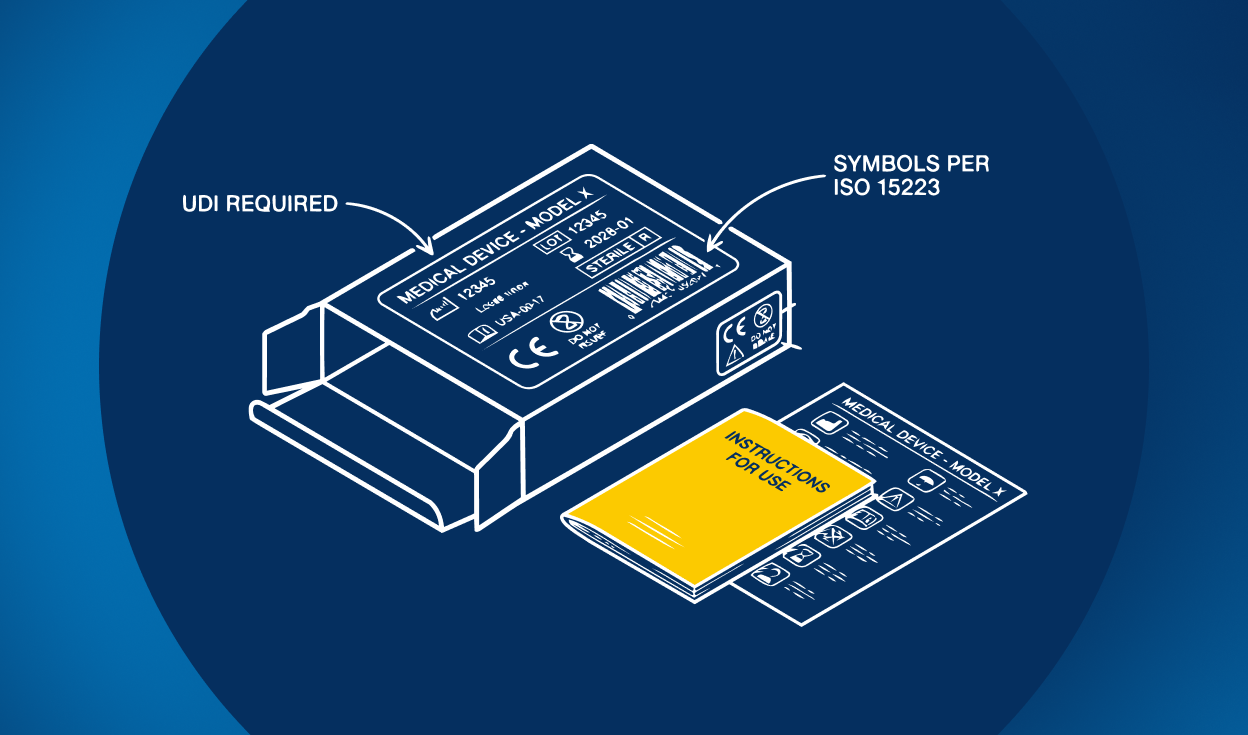
Table of contents


Medical device labeling is a critical part of regulatory compliance, patient safety, and product traceability. It goes far beyond packaging text — it connects regulatory data, product identification, and real-world use.
As global regulations evolve, particularly with FDA requirements, EU MDR, and UDI systems, manufacturers must ensure labeling is accurate, consistent, and fully auditable across all formats — from packaging and IFUs to digital labeling.
This guide explains what medical device labeling includes, the key regulatory requirements, and how to build a compliant labeling process.
What is a medical device?
Definitions of medical devices by the US Food and Drug Administration (FDA) and European Union (EU) are similar in many aspects and clearly outlined in respective labeling regulations.
In the US, medical devices are regulated by the FDA’s Center for Devices and Radiological Health [1] (CDRH), and applies to “firms who manufacture, repackage, relabel, and/or import medical devices sold in the United States.” Requirements are stipulated in Chapter V of the Food, Drug and Cosmetics Act, [2] and general labeling requirements are provided in 21 CFR Part 801. [3]
In the EU, the definition is laid down in “Regulation (EU) 2017/745 on Medical Devices” [4] (MDR) and the “Regulation (EU) 2017/746 on In Vitro Diagnostic Medical Devices.” [5]
In essence, both encompass instruments, machines and appliances, implants, reagents, materials, and software (see FDA here (What are examples of Software as a Medical Device?))[6] for medical purposes. The approaches they take are risk-based, classifying medical devices in risk classes. Two key differences should be kept in mind here, however.
What is medical device labeling?
Medical device labeling refers to all information provided with a device to ensure its safe and effective use.
This includes:
- Labels on packaging and devices
- Instructions for Use (IFU)
- Symbols and safety information
- Digital or electronic labeling (e-labeling)
- Accompanying regulatory documentation
Labeling is not just a regulatory requirement — it is a core safety mechanism that ensures healthcare professionals and patients use devices correctly.
Why medical device labeling requirements matter
Medical device labeling plays a central role in:
- Patient safety — incorrect or missing information can lead to misuse
- Regulatory compliance — required for FDA, MDR, and global approvals
- Traceability — essential for UDI and post-market surveillance
- Market access — non-compliant labeling can delay or block product launches
- Recall prevention — labeling errors are a major cause of product recalls
Even small inconsistencies — such as incorrect symbols, missing warnings, or mismatched data — can create significant compliance risks.
FDA and EU pathways
Firstly, both pharmaceuticals and medical devices are within the remit of the FDA. In the EU, the European Medicines Agency (EMA) plays a supportive role in the regulation of medical devices, but if a medicine itself performs the principle action, [7] the product is deemed to be a “medicinal product that includes a medical device”. Both medicine and device then fall within EU pharmaceutical legislation (i.e. require market authorization as a medicine), although the device itself may still require a conformity assessment.
For medical devices, recognized notified bodies [8] designated by each EU country assess product conformity. If they conform, the medical device receives its CE (conformity assessment) mark, which becomes part of the product labeling. The medical device labeling itself is an integral part of the assessment.
Secondly, the FDA takes a “predicate” approach for Class I (low risk) and Class II (moderate risk) medical devices, with a simplified path to market – so-called 510(k) submission [9] – with Class III devices requiring pre-market approval.
FDA and EU labeling requirements for medical device
While medical device labeling standards may be relatively simple for a manual toothbrush, labeling requirements for medical devices in high-risk classes are complex and stringently defined. Some general medical device labeling alignments stand out for safe operation and identification, however. These include the name and trade name of the device, information pertaining to the manufacturer, intended use, lot and serial numbers, the use of internationally recognized symbols, instructions for use, residual risks, and many other medical device labeling specifics.
In terms of electronic labeling (e-labeling), however, the EU MDR requires companies that have a website – and most do – to provide specific up-to-date labeling information online.
Although ISO/IEC standards in themselves do not constitute law, standards form an integral part of regulatory compliance, especially the requirement to have quality management systems in place. For example, EN ISO/IEC 17000 conformity standards and accreditation are paramount for EU/EEA CE marking by notified bodies, and the FDA is in the process of harmonizing [10] its quality system (QS; 21 CFR Part 820) regulation with ISO 13485:2016.
Why labeling is important:
- Patient safety and use (by patients and medical professionals) through instructions on proper use, handling and risks involved.
- Traceability of medical devices is paramount. The FDA and EU both have labeling requirements to include a Unique Device Identifier (UDI) on some classes of medical devices
- Compliance is jeopardized by incorrect or misleading labeling
- Legal responsibilities are implicitly defined in labeling (e.g. if instructions for use are not followed by a user, the manufacturer is less likely to be liable for any misuse).
- Learning and training is implicit in labeling (and other documentation)
How to build a compliant medical device labeling workflow
Step-by-step process:
- Define approved source content (regulatory master data)
- Map regulatory requirements by market (FDA, MDR, global)
- Assign UDI and traceability data
- Create labeling and artwork files
- Verify text, symbols, and identifiers
- Conduct regulatory, QA, and stakeholder review
- Approve with full audit trail
- Maintain change control across updates
Without a structured workflow, teams face:
- Version confusion
- Delays in approvals
- Increased compliance risk
4 tips on the road to best practices for medical device label requirements
1. Compliance
Make sure you have the systems, people and resources in place to understand, formulate and communicate regulatory requirements and quality standards. This includes practical requirements (e.g. font size, use of symbols, etc.).
2. Workflows and automation
Have clearly defined workflows and automate processes with software for labeling creation and updates. Processes must be replicable, collaborative, and promote accountability through audit trails.
3. Manage risk
Assess and understand what actions are high risk, and where errors might occur (e.g. manual proofreading of labeling is high-risk)
4. Align paper and online labeling
Electronic labeling (e-labeling) is a reality, even if the pace of implementation varies from country to country. Your software and workflows must be able to align paper and online formats for labeling creation and especially for updates.
How TVT, the document comparison software, supports labeling compliance
Document comparison software, like TVT minimizes the risk of error by verifying text, spelling, artwork, tables and barcodes in labeling, packaging and documentation before getting printed or published. TVT is used not only by leading medical device companies, but also by regulatory authorities.
You can build your proofreading and verification workflows around TVT, allowing you to work faster and quickly find mistakes in labeling and packaging (e.g. by comparing print proofs with an approved original labeling document for deviations). Online information can be easily loaded and compared with other formats which is a key component of both the EU MDR and e-labeling initiatives mentioned above. This is important when you have patient information leaflets and other documentation in online and paper format.
If you would like to find out how TVT can help you verify your labeling, save time and reduce the risk of non-compliance, contact us below.
What is medical device labeling?
Medical device labeling includes all information provided with a device to ensure safe and effective use, including packaging labels, IFUs, symbols, and digital instructions.
What are UDI requirements for medical devices?
UDI requirements mandate that devices include a unique identifier for traceability, enabling better monitoring, recall management, and regulatory oversight.
What is the difference between FDA and MDR labeling requirements?
FDA labeling focuses on safe use and compliance within the U.S., while MDR adds stricter requirements for traceability, language localization, and lifecycle documentation across the EU.
What labeling errors cause recalls?
Common causes include incorrect or missing UDI, inconsistent data across materials, missing warnings, and translation errors.
How can manufacturers improve labeling compliance?
By implementing structured workflows, ensuring data consistency, and using automated verification tools to detect errors before approval.
References
1. FDA overview of device regulation:
www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance/overview-device-regulation
2. FD&C Act Chapter V: Drugs and Devices:
https://www.fda.gov/regulatory-information/federal-food-drug-and-cosmetic-act-fdc-act/fdc-act-chapter-v-drugs-and-devices
3. FDA Device Labeling:
https://www.fda.gov/medical-devices/overview-device-regulation/device-labeling
4. Overview medical devices EU: Medical devices https://www.ema.europa.eu/en/human-regulatory/overview/medical-devices
5. EU in vitro diagnostic medical devices regulation 2017/746:
https://eur-lex.europa.eu/eli/reg/2017/746/oj
6. FDA examples of software as a medical device:
https://www.fda.gov/medical-devices/software-medical-device-samd/what-are-examples-software-medical-device
7. Overview medical devices EU: Medical devices https://www.ema.europa.eu/en/human-regulatory/overview/medical-devices
8. About notified bodies in the EU/EEA:
https://single-market-economy.ec.europa.eu/single-market/goods/building-blocks/notified-bodies_en
9. FDA Premarket Notification 510(k):
https://www.fda.gov/medical-devices/premarket-submissions-selecting-and-preparing-correct-submission/premarket-notification-510k#intro
10. FDA harmonization of quality system regulations:
https://www.fda.gov/medical-devices/postmarket-requirements-devices/quality-system-qs-regulationmedical-device-good-manufacturing-practices